BioNeMo-SCDL: Single Cell Data Loading for Scalable Training of Single Cell Foundation Models.
Package Overview
BioNeMo-SCDL provides an independent pytorch-compatible dataset class for single cell data with a consistent API. BioNeMo-SCDL is developed and maintained by NVIDIA. This package can be run independently from BioNeMo. It improves upon simple AnnData-based dataset classes in the following ways:
- A consistent API across input formats that is promised to be consistent across package versions.
- Improved performance when loading large datasets. It allows for loading and fast iteration of large datasets.
- Ability to use datasets that are much, much larger than memory. This is because the datasets are stored in a numpy memory-mapped format.
- Additionally, conversion of large (significantly larger than memory) AnnData files into the SCDL format.
- [Future] Full support for ragged arrays (i.e., datasets with different feature counts; currently only a subset of the API functionality is supported for ragged arrays).
- [Future] Support for improved compression.
BioNeMo-SCDL's API resembles that of AnnData, so code changes are minimal.
In most places a simple swap from an attribute to a function is sufficient (i.e., swapping data.n_obs for data.number_of_rows()).
Installation
This package can be installed with
pip install bionemo-scdl
Usage
Getting example data
Here is how to process an example dataset from CellxGene with ~25,000 cells:
Download "https://datasets.cellxgene.cziscience.com/97e96fb1-8caf-4f08-9174-27308eabd4ea.h5ad" to hdf5s/97e96fb1-8caf-4f08-9174-27308eabd4ea.h5ad
Loading a single cell dataset from an H5AD file
from bionemo.scdl.io.single_cell_memmap_dataset import SingleCellMemMapDataset
data = SingleCellMemMapDataset(
"97e_scmm", "hdf5s/97e96fb1-8caf-4f08-9174-27308eabd4ea.h5ad"
)
This creates a SingleCellMemMapDataset that is stored at 97e_scmm in large, memory-mapped arrays
that enables fast access of datasets larger than the available amount of RAM on a system.
If the dataset is large, the AnnData file can be lazy-loaded and then read in based on chunks of rows in a paginated manner. This can be done by setting the parameters when instantiating the SingleCellMemMapDataset:
paginated_load_cutoff, which sets the minimal file size in megabytes at which an AnnData file will be read in in a paginated manner.load_block_row_size, which is the number of rows that are read into memory at a given time.
Loading raw.X vs .X from the anndata file
By default, SCDL will load the data from the raw.X in the anndata file. If using the .X is desired, set use_X_not_raw = True
during the dataset creation:
from bionemo.scdl.io.single_cell_memmap_dataset import SingleCellMemMapDataset
data = SingleCellMemMapDataset(
"97e_scmm", "hdf5s/97e96fb1-8caf-4f08-9174-27308eabd4ea.h5ad"
)
Interrogating single cell datasets and exploring the API
data.number_of_rows()
## 25382
data.number_of_variables()
## [34455]
data.number_of_values()
## 874536810
data.number_nonzero_values()
## 26947275
Saving SCDL (Single Cell DataLoader) datasets to disk
When you open a SCDL dataset, you must choose a path where the backing data structures are stored. However, these structures are not guaranteed to be in a valid serialized state during runtime.
Calling the save method guarantees the on-disk object is in a valid serialized
state, at which point the current python process can exit, and the object can be
loaded by another process later.
data.save()
Loading SCDL datasets from a SCDL archive
When you're ready to reload a SCDL dataset, just pass the path to the serialized data:
reloaded_data = SingleCellMemMapDataset("97e_scmm")
Using SCDL datasets in model training
SCDL implements the required functions of the PyTorch Dataset abstract class.
Tokenization
A common use case for the single-cell dataloader is tokenizing data using a predefined vocabulary with a defined
tokenizer function. These features (that in anndata are stored in .var) can be accessed with return_var_features
and setting var_feature_names to the desired feature names. Similarly, row-wise features (that are in the .obs,)
can be accessed with return_obs_features and setting obs_feature_names.
import numpy as np
ds = SingleCellMemMapDataset("97e_scmm")
index = 0
values, var_feature_ids, obs_feature_ids = ds.get_row(
index,
return_var_features=True,
var_feature_names=["feature_id"],
return_obs_features=True,
obs_feature_names=["cell_line"],
)
assert (
len(var_feature_ids) == 1 and len(obs_feature_ids) == 1
) # we expect feature_ids to be a list containing one np.array with the row's feature ids
gene_data, col_idxs = np.array(values[0]), np.array(values[1])
tokenizer_function = lambda x, y, z, w: x
tokenizer_function(gene_data, col_idxs, var_feature_ids[0], obs_feature_ids[0])
Observed (.obs) features
Observed (row-level, per-cell) features can be accessed using the .obs_features() method on your dataset instance. This
method allows you to retrieve per-cell metadata stored in the .obs of underlying AnnData files, either as a single dictionary
(if your SCDL archive came from a single AnnData file) or as a list of dictionaries (if multiple input files were concatenated and their .obs columns differ).
You can use integer indexing to get the features for a single cell, or slicing to get features for a range of cells:
# Get .obs features for cells 5 through 9
df = data.obs_features()[5:10]
# Get .obs features for cell 3
row = data.obs_features()[3]
Loading directly with PyTorch-compatible DataLoaders
You can use PyTorch-compatible DataLoaders to load batches of data from a SCDL class.
With a batch size of 1, this can be run without a collation function. With a batch size
greater than 1, there is a collation function (collate_sparse_matrix_batch) that will
collate several sparse arrays into the CSR (Compressed Sparse Row) PyTorch tensor format.
from torch.utils.data import DataLoader
from bionemo.scdl.util.torch_dataloader_utils import collate_sparse_matrix_batch
## Mock model: you can remove this and pass the batch to your own model in actual code.
model = lambda x: x
dataloader = DataLoader(
data, batch_size=8, shuffle=True, collate_fn=collate_sparse_matrix_batch
)
n_epochs = 2
for e in range(n_epochs):
for batch in dataloader:
model(batch)
Data Type Casting
SCDL lets you control both storage size and numerical precision by specifying the data type for values loaded from AnnData .X. Supported types include "uint8", "uint16", "uint32", "uint64", "float16", "float32", and "float64". Choosing a smaller type (like "uint8" or "float16") results in more compact storage, while selecting a higher-precision type (such as "float64") uses more space but preserves maximum accuracy. You set the data type at the time of dataset creation from an AnnData file using:
from bionemo.scdl.io.single_cell_memmap_dataset import SingleCellMemMapDataset
data = SingleCellMemMapDataset(
"97e_scmm", "hdf5s/97e96fb1-8caf-4f08-9174-27308eabd4ea.h5ad", data_dtype="uint64"
)
SCDL checks for minimal loss when doing this. The amount of tolerated loss in the data is set through the data_dtype_tolerance parameter.
Changing data dtype after creation (in-place)
If you need to change the on-disk data dtype after a dataset has been created, you can cast it in place:
from bionemo.scdl.io.single_cell_memmap_dataset import SingleCellMemMapDataset
ds = SingleCellMemMapDataset("97e_scmm")
ds.cast_data_to_dtype("float64") # or "uint16", "float32", etc.
# Optionally reopen to verify
reloaded = SingleCellMemMapDataset("97e_scmm")
assert reloaded.dtypes["data.npy"] == "float64"
Notes:
- Casting is done in place and updates the on-disk arrays and dtype registry.
- Avoid mixing integer and floating‑point families across datasets you plan to concatenate; SCDL raises an error when families differ.
Examples
The examples directory contains various examples for utilizing SCDL.
Converting existing CellxGene data to SCDL
If there are multiple AnnData files, they can be converted into a single SingleCellMemMapDataset. If the hdf5 directory has one or more AnnData files, the SingleCellCollection class crawls the filesystem to recursively find AnnData files (with the h5ad extension).
To convert existing AnnData files, you can either write your own script using the SCDL API or utilize the convenience script convert_h5ad_to_scdl.
During dataset concatenation, it is assumed that all of the data types are either floats or ints, and all of the entries are upscaled to the largest data size. If there is a combination of floats and ints when concatenating the data, an error is thrown.
To convert multiple files with a given data format, the user can run:
convert_h5ad_to_scdl --data-path hdf5s --save-path example_dataset [--data-dtype float64 --paginated_load_cutoff 10_000 --load-block-row-size 1_000_000 --use-X-not-raw]
Runtimes with SCDL
The runtime is examined on the Tahoe 100M dataset, which contains over 100 million rows. On this dataset, there is either a 12× or 53× speedup depending on the machine used.
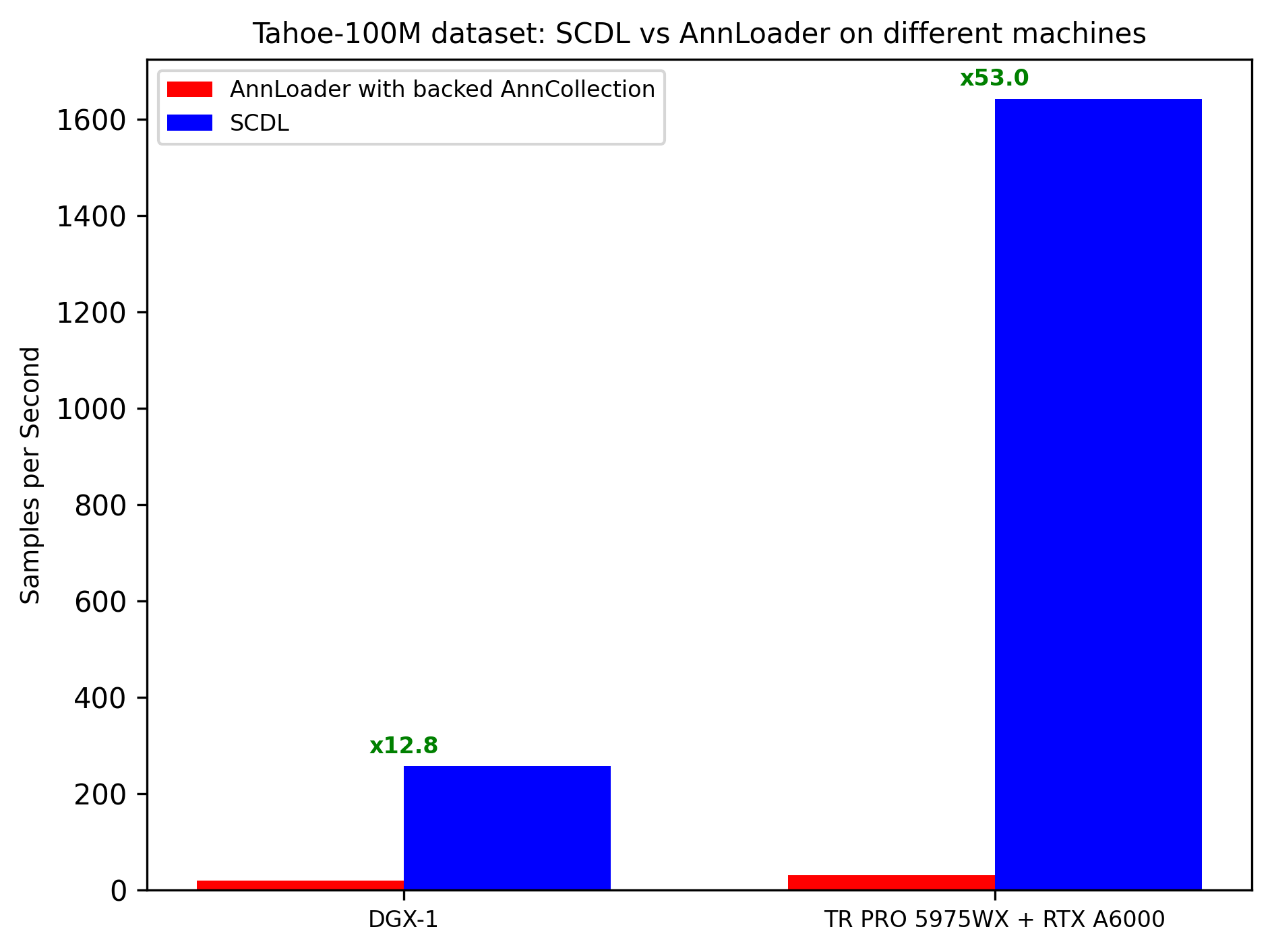
To replicate this on your machine, see: Tahoe 100M Profiling section.
Using Neighbor Information in Single Cell Datasets
SCDL now supports loading and utilizing neighbor information from AnnData objects. This is particularly useful for tasks that require knowledge of cell neighborhoods, trajectory analysis, or spatial relationships.
Neighbor Data Structure in AnnData
The neighbor functionality reads neighbor information from the .obsp (observations pairwise) attribute of the AnnData object and converts it from sparse matrix format into SCDL's memory-mapped format for efficient access:
- Input Location:
adata.obsp[neighbor_key](default key is'next_cell_ids') - Input Format: Sparse matrix (scipy.sparse format, typically CSR - Compressed Sparse Row)
- SCDL Processing: Converts sparse neighbor data into memory-mapped arrays during dataset creation
- Dimensions:
[n_cells × n_cells]adjacency matrix - Values: Weights/distances (e.g., pseudotime values, spatial distances, similarity scores)
- Non-zero entries: Indicate neighbor relationships
Example - Generating Neighbor Data from Trajectory Analysis:
import scanpy as sc
import numpy as np
from scipy.sparse import csr_matrix
# After computing pseudotime with your preferred method (e.g., DPT, Monocle, etc.)
# adata.obs['pseudotime'] contains pseudotime values for each cell
# Assuming you define a function create_pseudotime_neighbors() to find k nearest neighbors in pseudotime space and store as sparse matrix
# Create and store neighbor matrix
neighbor_matrix = create_pseudotime_neighbors(adata.obs["pseudotime"])
adata.obsp["next_cell_ids"] = neighbor_matrix
Loading a Dataset with Neighbor Support
from bionemo.scdl.io.single_cell_memmap_dataset import (
SingleCellMemMapDataset,
NeighborSamplingStrategy,
)
# Load dataset with neighbor support
data = SingleCellMemMapDataset(
"dataset_path",
"path/to/anndata.h5ad",
load_neighbors=True, # Enable neighbor functionality
neighbor_key="next_cell_ids", # Key in AnnData.obsp containing neighbor information
neighbor_sampling_strategy=NeighborSamplingStrategy.RANDOM, # Strategy for sampling neighbors
fallback_to_identity=True, # Use cell itself as neighbor when no neighbors exist
)
Accessing Neighbor Data
SCDL provides several methods to access and utilize neighbor information:
# Get neighbor indices for a specific cell
neighbor_indices = data.get_neighbor_indices_for_cell(cell_index)
# Get neighbor weights (if available)
neighbor_weights = data.get_neighbor_weights_for_cell(cell_index)
# Sample a neighbor according to the configured strategy
neighbor_index = data.sample_neighbor_index(cell_index)
Example Usage in Contrastive Learning:
# Contrastive Learning - Compare cells with their neighbors
for cell_index in range(len(data)):
# Get current cell and its neighbor
current_cell_data, _ = data.get_row(cell_index)
neighbor_index = data.sample_neighbor_index(cell_index)
neighbor_cell_data, _ = data.get_row(neighbor_index)
# Use in contrastive loss
current_embedding = model.encode(current_cell_data)
neighbor_embedding = model.encode(neighbor_cell_data)
contrastive_loss = compute_contrastive_loss(current_embedding, neighbor_embedding)
Future Work and Roadmap
SCDL is currently in public beta. In the future, expect improvements in data compression and data loading performance.
LICENSE
BioNeMo-SCDL has an Apache 2.0 license, as found in the LICENSE file.
Contributing
Please follow the guidelines for contributions to the BioNeMo Framework.
To contribute to SCDL, we recommend installing additional dependencies for development and installing the SCDL package from source.
git clone https://github.com/NVIDIA/bionemo-framework.git
cd bionemo-framework/sub-packages/bionemo-scdl
pip install -e ".[test]"
Tests
SCDL has its own tests. To run these tests, assuming you have pytest installed:
python -m pytest
To run a specific test:
python -m pytest tests/test_<test name>.py
Troubleshooting
-
Mixed data types at concat (ValueError: mix of int and float dtypes)
-
Cause: attempting to concatenate datasets whose data arrays are from different dtype families (e.g., one integer, one floating‑point).
-
Fixes:
- Recast all input archives to a common dtype family. You can do this in place with
ds.cast_data_to_dtype("float32"), then rerun concatenation. - Alternatively, rebuild inputs using
convert_h5ad_to_scdl --data-dtype <dtype>so they share the same family.
- Recast all input archives to a common dtype family. You can do this in place with
-
OOM during dataset instantiation or concatenation from h5ad files.
-
Cause: Likely due to overly large chunks of the anndata file being read into memory.
-
Fixes: Set a lower paginated_load_cutoff, load_block_row_size, or number of workers during concatenation.
-
Slow DataLoader throughput when returning rich Python structures
-
Cause: returning dicts or strings from
Dataset/collate_fnprevents fast vectorized collation. -
Fixes:
- Return tensors only; prefer a tuple
(X, idx)and gather.obsinside the model from a pre‑encoded tensor aligned to row order.
- Return tensors only; prefer a tuple
-
Downcasting warnings (data precision loss)
-
Cause: requested
data_dtypeis narrower than the source values allow. - Fixes:
- Choose a wider dtype (e.g.,
float32/float64, or a larger unsigned int), or raisedata_dtype_toleranceas appropriate.
- Choose a wider dtype (e.g.,