Variational Quantum Eigensolver¶
A common application of the Variational Quantum Eigensolver (VQE) algorithm is to compute the ground state energy of a molecular system. The code below demonstrates how to perform classical preprocessing for a \(H_2\) molecule (i.e. obtain the integrals from a Hartree-Fock computation to build the molecular Hamiltonian), prepare the initial Hartree-Fock state on the quantum register, add the parameterized UCCSD ansatz to the kernel, and select the COBYLA optimizer. We are then ready to call
cudaq:vqe to estimate the minimum energy of the system.
[1]:
# !pip install openfermionpyscf matplotlib scipy
[2]:
import cudaq
import matplotlib.pyplot as plt
from scipy.optimize import minimize
import numpy as np
np.random.seed(42)
cudaq.set_target("nvidia")
The problem of interest here is a chain of hydrogen atoms seperated along the z-axis at a fixed interval called the bond distance.
The interatomic electrostatic forces due to the electrons and protons and the shielding by the neutrons creates a chemical system whose energy can be minimised to find a stable configuration.
Let us first begin by defining the molecule and other metadata about the problem.
[3]:
# Number of hydrogen atoms.
hydrogen_count = 2
# Distance between the atoms in Angstroms.
bond_distance = 0.7474
# Define a linear chain of Hydrogen atoms
geometry = [('H', (0, 0, i * bond_distance)) for i in range(hydrogen_count)]
molecule, data = cudaq.chemistry.create_molecular_hamiltonian(
geometry, 'sto-3g', 1, 0)
electron_count = data.n_electrons
qubit_count = 2 * data.n_orbitals
We now generate a Unitary Coupled-Cluster Singles and Doubles (UCCSD) ansatz from the template provided by CUDA Quantum.
[1]:
kernel, angles = cudaq.make_kernel(list)
qubits = kernel.qalloc(qubit_count)
# Prepare the Hartree Fock State.
kernel.x(qubits[0])
kernel.x(qubits[1])
# Adds parameterized gates based on the UCCSD ansatz.
kernel.apply_call(cudaq.kernels.uccsd, qubits, angles, electron_count,
qubit_count)
parameter_count = cudaq.kernels.uccsd_num_parameters(electron_count,
qubit_count)
-1.1371745102369863
Using CUDA Quantum Optimizers¶
We use the builtin optimizers within CUDA Quantum for the minimization procedure.
[5]:
optimizer = cudaq.optimizers.COBYLA()
energy, parameters = cudaq.vqe(kernel,
molecule,
optimizer,
argument_mapper=lambda parameters:
(parameters, qubit_count, electron_count),
parameter_count=parameter_count)
print(energy)
-1.1371756094989427
Integration with Third-Party Optimizers¶
We can also integrate popular libraries like scipy with CUDA Quantum.
[6]:
# Define a function to minimize
def to_minimize(theta):
exp_val = cudaq.observe(kernel, molecule, theta).expectation()
return exp_val
exp_vals = []
def callback(xk):
exp_vals.append(to_minimize(xk))
# Initial variational parameters.
x0 = np.random.uniform(low=0, high=2 * np.pi, size=parameter_count)
# Use the scipy optimizer to minimize the function of interest
result = minimize(to_minimize,
x0,
method='COBYLA',
callback=callback,
options={'maxiter': 40})
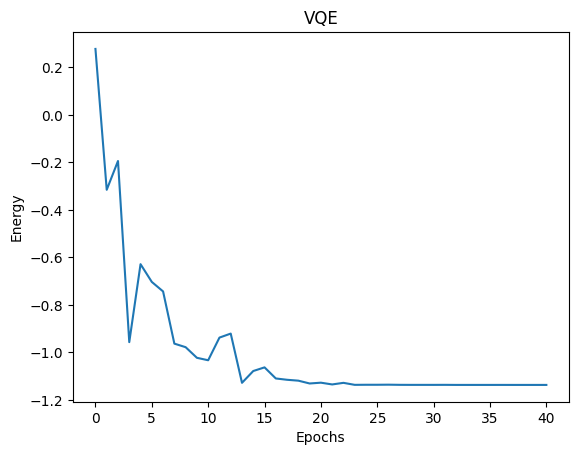
plt.plot(exp_vals)
plt.xlabel('Epochs')
plt.ylabel('Energy')
plt.title('VQE')
plt.show()