Note
Go to the end to download the full example code.
Variable-Cell FIRE2 Optimization with LJ Potential#
This example demonstrates joint optimization of atomic positions and cell parameters using the coupled FIRE2 PyTorch adapter (Guenole et al., 2020).
Compared to FIRE (07_fire_variable_cell.py), FIRE2:
Assumes unit mass (no
masses/pack_masses_with_cellneeded)Requires
batch_idx(all zeros for this single-system example)Has simpler state:
alpha,dt,nsteps_incHyperparameters are Python scalars
The workflow is otherwise the same:
align_cell() - Transform cell to upper-triangular form
LJ energy/forces/virial - Compute interatomic interactions
Virial -> Stress -> Cell Force - Convert virial to cell driving force
fire2_step_coord_cell() - Coupled coordinate/cell FIRE2 update
We optimize an FCC argon crystal under external pressure.
from __future__ import annotations
import matplotlib.pyplot as plt
import numpy as np
import torch
import warp as wp
from _dynamics_utils import (
EPSILON_AR,
SIGMA_AR,
MDSystem,
create_fcc_lattice,
pressure_ev_per_a3_to_gpa,
pressure_gpa_to_ev_per_a3,
virial_to_stress,
)
from nvalchemiops.dynamics.utils import (
align_cell,
compute_cell_volume,
stress_to_cell_force,
wrap_positions_to_cell,
)
from nvalchemiops.torch import fire2_step_coord_cell
# LJ cutoff for argon
CUTOFF = 2.5 * SIGMA_AR # ~8.5 Å
# ==============================================================================
# Main Example
# ==============================================================================
wp.init()
device = "cuda:0" if wp.is_cuda_available() else "cpu"
print(f"Using device: {device}")
Using device: cuda:0
Create Initial System#
n_cells = 3 # 3x3x3 = 108 atoms
a_initial = 5.5 # Å (slightly expanded from equilibrium ~5.26 Å)
positions_np, cell_np = create_fcc_lattice(n_cells, a_initial)
num_atoms = len(positions_np)
# Target external pressure (positive = compression)
target_pressure_gpa = 0.01
target_pressure = pressure_gpa_to_ev_per_a3(target_pressure_gpa)
print(f"System: {num_atoms} atoms in {n_cells}\u00b3 FCC lattice")
print(f"Initial lattice constant: {a_initial:.3f} \u00c5")
print(f"Initial density: {num_atoms / np.linalg.det(cell_np):.4f} atoms/\u00c5\u00b3")
print(f"Target external pressure: {target_pressure_gpa:.3f} GPa")
print(f"LJ parameters: \u03b5 = {EPSILON_AR:.4f} eV, \u03c3 = {SIGMA_AR:.2f} \u00c5")
System: 108 atoms in 3³ FCC lattice
Initial lattice constant: 5.500 Å
Initial density: 0.0240 atoms/ų
Target external pressure: 0.010 GPa
LJ parameters: ε = 0.0104 eV, σ = 3.40 Å
Initialize System#
# Create Warp arrays
positions = wp.array(positions_np, dtype=wp.vec3d, device=device)
cell = wp.array(cell_np.reshape(1, 3, 3), dtype=wp.mat33d, device=device)
# Create MD system for force computation
md_system = MDSystem(
positions=positions_np,
cell=cell_np,
epsilon=EPSILON_AR,
sigma=SIGMA_AR,
cutoff=CUTOFF,
skin=0.5,
switch_width=1.0, # Smooth cutoff for optimization
device=device,
)
Initialized MD system with 108 atoms
Cell: 16.50 x 16.50 x 16.50 Å
Cutoff: 8.50 Å (+ 0.50 Å skin)
LJ: ε = 0.0104 eV, σ = 3.40 Å
Device: cuda:0, dtype: <class 'numpy.float64'>
Units: x [Å], t [fs], E [eV], m [eV·fs²/Ų] (from amu), v [Å/fs]
Step 1: Align Cell#
--- Step 1: Align cell to upper-triangular form ---
Aligned cell:
[[16.5 0. 0. ]
[ 0. 16.5 0. ]
[ 0. 0. 16.5]]
Step 2: Create Optimizer Tensors#
print("\n--- Step 2: Create optimizer tensors ---")
torch_device = torch.device(device)
positions_t = wp.to_torch(positions)
cell_t = wp.to_torch(cell).reshape(1, 3, 3)
velocities_t = torch.zeros_like(positions_t)
cell_velocities_t = torch.zeros_like(cell_t)
batch_idx_t = torch.zeros(num_atoms, dtype=torch.int32, device=torch_device)
print(f"Optimizer state: {num_atoms} atoms + 1 cell")
--- Step 2: Create optimizer tensors ---
Optimizer state: 108 atoms + 1 cell
Step 3: FIRE2 Parameters#
# Per-system state arrays (shape (1,) for single system)
alpha = torch.tensor([0.09], dtype=torch.float64, device=torch_device)
dt = torch.tensor([0.005], dtype=torch.float64, device=torch_device)
nsteps_inc = torch.zeros(1, dtype=torch.int32, device=torch_device)
cell_force_scale = 1.0
# Scratch buffers (shape (1,) for single system)
vf = torch.zeros(1, dtype=torch.float64, device=torch_device)
v_sumsq = torch.zeros(1, dtype=torch.float64, device=torch_device)
f_sumsq = torch.zeros(1, dtype=torch.float64, device=torch_device)
max_norm_buf = torch.zeros(1, dtype=torch.float64, device=torch_device)
# Scratch arrays for unpack/stress/volume
cell_force_scratch = wp.empty(1, dtype=wp.mat33d, device=device)
volume_scratch = wp.empty(1, dtype=wp.float64, device=device)
Step 4: Optimization Loop#
max_steps = 1000
force_tol = 1e-4 # Convergence: max atomic force component
pressure_tol_gpa = 0.03
log_interval = 100
check_interval = 50
# History for plotting
energy_hist = []
max_force_hist = []
volume_hist = []
pressure_hist = []
lattice_const_hist = []
print("\n--- Step 4: Variable-cell FIRE2 optimization ---")
print(f"Force tolerance: {force_tol:.1e}")
print(f"Stress tolerance: {pressure_tol_gpa:.2e} GPa")
print(
"FIRE2 defaults: delaystep=60, dtgrow=1.05, alpha0=0.09, "
"maxstep=0.1, cell_force_scale=1.0"
)
print("=" * 90)
print(
f"{'Step':>6} {'Energy':>12} {'max|F|':>10} {'Volume':>10} "
f"{'|stress|':>10} {'a (Å)':>10}"
)
print("=" * 90)
converged = False
for step in range(max_steps):
# Update MD system with current geometry
wp.copy(md_system.wp_positions, positions)
md_system.update_cell(cell)
# Wrap positions into cell (important for PBC consistency)
wrap_positions_to_cell(
positions=md_system.wp_positions,
cells=md_system.wp_cell,
cells_inv=md_system.wp_cell_inv,
device=device,
)
wp.copy(positions, md_system.wp_positions)
# Compute LJ forces and virial
energies, forces, virial = md_system.compute_forces_virial()
# Convert virial to stress with external pressure contribution
stress = virial_to_stress(virial, md_system.wp_cell, target_pressure, device)
# Convert stress to cell force (for optimization)
compute_cell_volume(md_system.wp_cell, volumes=volume_scratch, device=device)
stress_to_cell_force(
stress,
md_system.wp_cell,
volume=volume_scratch,
cell_force=cell_force_scratch,
keep_aligned=True,
device=device,
)
# Coupled FIRE2 step on coordinates and cell.
fire2_step_coord_cell(
positions=positions_t,
velocities=velocities_t,
forces=wp.to_torch(forces),
cell=cell_t,
cell_velocities=cell_velocities_t,
cell_force=wp.to_torch(cell_force_scratch).reshape(1, 3, 3),
batch_idx=batch_idx_t,
alpha=alpha,
dt=dt,
nsteps_inc=nsteps_inc,
vf=vf,
v_sumsq=v_sumsq,
f_sumsq=f_sumsq,
max_norm=max_norm_buf,
cell_force_scale=cell_force_scale,
)
# Check convergence and log only at intervals
if step % check_interval == 0 or step == max_steps - 1:
wp.synchronize()
force_max = np.max(np.abs(forces.numpy()))
max_force = force_max
total_energy = float(energies.numpy().sum())
compute_cell_volume(md_system.wp_cell, volumes=volume_scratch, device=device)
volume = float(volume_scratch.numpy()[0])
stress_np = stress.numpy()[0]
stress_gpa = pressure_ev_per_a3_to_gpa(0.5 * (stress_np + stress_np.T))
stress_residual_gpa = np.linalg.svd(stress_gpa, compute_uv=False).max()
# Effective lattice constant (cube root of volume per atom * 4 for FCC)
lattice_const = (volume / num_atoms * 4) ** (1 / 3)
energy_hist.append(total_energy)
max_force_hist.append(max_force)
volume_hist.append(volume)
pressure_hist.append(stress_residual_gpa)
lattice_const_hist.append(lattice_const)
if step % log_interval == 0 or step == max_steps - 1:
print(
f"{step:>6d} {total_energy:>12.6f} {max_force:>10.2e} {volume:>10.2f} "
f"{stress_residual_gpa:>10.4f} {lattice_const:>10.4f}"
)
if max_force < force_tol and stress_residual_gpa < pressure_tol_gpa:
print(
f"\nConverged at step {step} "
f"(max|F| = {max_force:.2e}, |stress| = {stress_residual_gpa:.2e} GPa)"
)
converged = True
break
--- Step 4: Variable-cell FIRE2 optimization ---
Force tolerance: 1.0e-04
Stress tolerance: 3.00e-02 GPa
FIRE2 defaults: delaystep=60, dtgrow=1.05, alpha0=0.09, maxstep=0.1, cell_force_scale=1.0
==========================================================================================
Step Energy max|F| Volume |stress| a (Å)
==========================================================================================
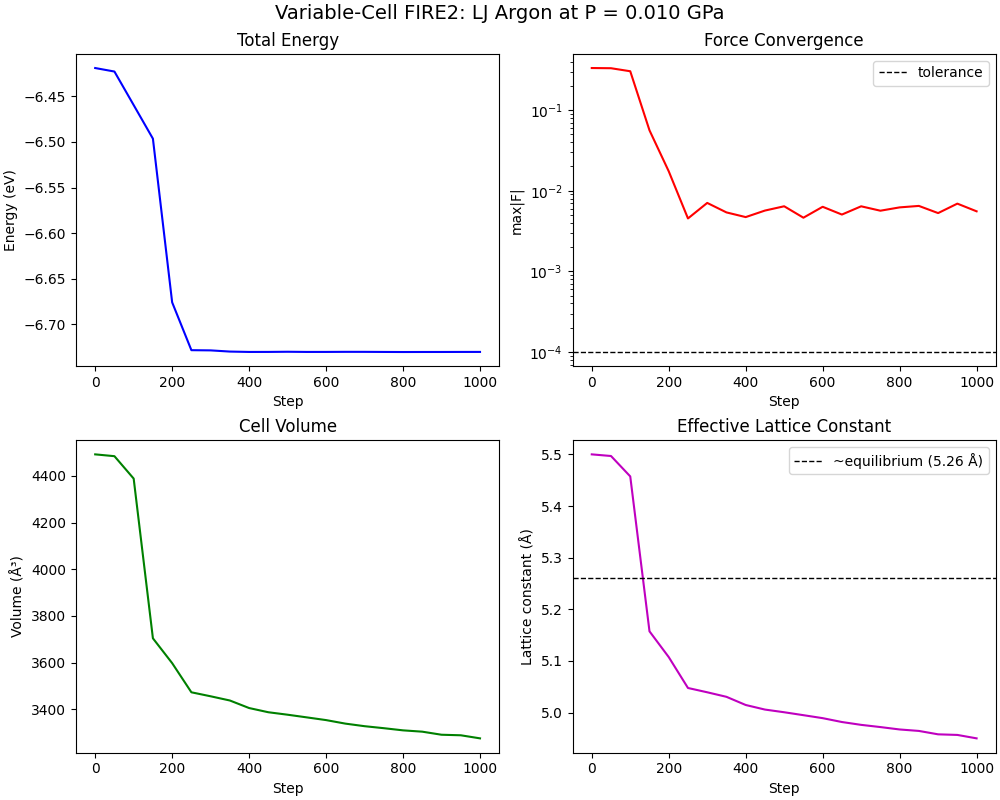
0 -8.410375 2.48e-16 4492.12 0.2434 5.5000
100 -8.412350 1.74e-15 4490.77 0.2431 5.4994
200 -8.560203 2.06e-15 4382.55 0.2132 5.4549
300 -8.811472 3.88e-15 4115.94 0.0997 5.3420
Converged at step 350 (max|F| = 4.16e-15, |stress| = 1.05e-02 GPa)
Final Results#
wp.synchronize()
final_pos = positions
final_cell = cell
wp.synchronize()
final_cell_np = final_cell.numpy()[0]
final_volume = np.linalg.det(final_cell_np)
final_density = num_atoms / final_volume
final_a = (final_volume / num_atoms * 4) ** (1 / 3)
print("\n" + "=" * 60)
print("FINAL RESULTS")
print("=" * 60)
print(f"Final cell:\n{final_cell_np}")
print(f"\nFinal volume: {final_volume:.2f} \u00c5\u00b3")
print(f"Final density: {final_density:.6f} atoms/\u00c5\u00b3")
print(f"Effective lattice constant: {final_a:.4f} \u00c5")
print(f"Target pressure: {target_pressure_gpa:.4f} GPa")
============================================================
FINAL RESULTS
============================================================
Final cell:
[[ 1.58083726e+01 0.00000000e+00 0.00000000e+00]
[-4.80813831e-19 1.58083726e+01 0.00000000e+00]
[-2.53923756e-18 -2.63596751e-19 1.58083726e+01]]
Final volume: 3950.59 ų
Final density: 0.027338 atoms/ų
Effective lattice constant: 5.2695 Å
Target pressure: 0.0100 GPa
Plot Convergence#
fig, axes = plt.subplots(2, 2, figsize=(10, 8), constrained_layout=True)
steps = np.arange(len(energy_hist)) * check_interval
# Energy
axes[0, 0].plot(steps, energy_hist, "b-", lw=1.5)
axes[0, 0].set_xlabel("Step")
axes[0, 0].set_ylabel("Energy (eV)")
axes[0, 0].set_title("Total Energy")
# Force convergence
axes[0, 1].semilogy(steps, max_force_hist, "r-", lw=1.5)
axes[0, 1].axhline(force_tol, color="k", ls="--", lw=1, label="tolerance")
axes[0, 1].set_xlabel("Step")
axes[0, 1].set_ylabel("max|F|")
axes[0, 1].set_title("Force Convergence")
axes[0, 1].legend()
# Volume
axes[1, 0].plot(steps, volume_hist, "g-", lw=1.5)
axes[1, 0].set_xlabel("Step")
axes[1, 0].set_ylabel("Volume (\u00c5\u00b3)")
axes[1, 0].set_title("Cell Volume")
# Lattice constant
axes[1, 1].plot(steps, lattice_const_hist, "m-", lw=1.5)
axes[1, 1].axhline(5.26, color="k", ls="--", lw=1, label="~equilibrium (5.26 \u00c5)")
axes[1, 1].set_xlabel("Step")
axes[1, 1].set_ylabel("Lattice constant (\u00c5)")
axes[1, 1].set_title("Effective Lattice Constant")
axes[1, 1].legend()
fig.suptitle(
f"Variable-Cell FIRE2: LJ Argon at P = {target_pressure_gpa:.3f} GPa", fontsize=14
)
plt.show()
Total running time of the script: (0 minutes 0.762 seconds)