Quantum-Selected Configuration Interaction (QSCI)¶
Quantum-Selected Configuration Interaction (QSCI) [1] is a quantum-classical hybrid algorithm designed for electronic structure calculations, particularly to compoute ground state and excited state energies of molecular systems. This method is one of the quantum subspace diagonalization (QSD). It is well-suited for NISQ-era quantum devices, focusing on using quantum resources efficiently while maintaining high accuracy.
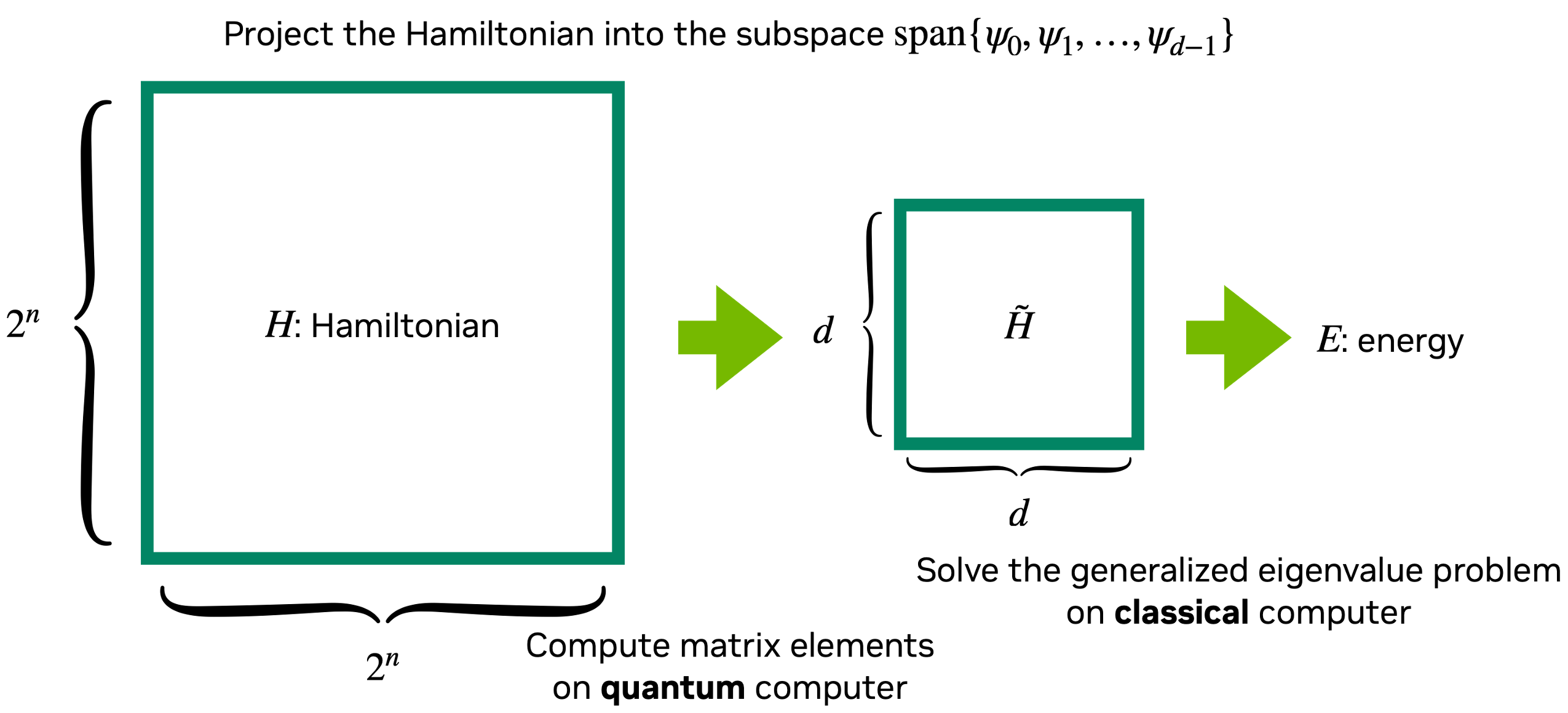
QSCI selects this vector space from a sampled computational basis.
0. Problem definition¶
Let’s define Hamiltonian of \(\mathrm{H_4}\).
[1]:
!pip install --quiet --break-system-packages openfermionpyscf==0.5
[2]:
import openfermion
import openfermionpyscf
from openfermion.transforms import jordan_wigner, get_fermion_operator
import cudaq
import numpy as np
if not hasattr(np, 'string_'):
np.string_ = np.bytes_
[3]:
# Number of hydrogen atoms.
hydrogen_count = 4
# Distance between the atoms in Angstroms.
bond_distance = 0.7474
# Define a linear chain of Hydrogen atoms
geometry = [("H", (0, 0, i * bond_distance)) for i in range(hydrogen_count)]
basis = "sto3g"
multiplicity = 1
charge = 0
molecule = openfermionpyscf.run_pyscf(
openfermion.MolecularData(geometry, basis, multiplicity, charge), run_fci=True
)
molecular_hamiltonian = molecule.get_molecular_hamiltonian()
fermion_hamiltonian = get_fermion_operator(molecular_hamiltonian)
qubit_hamiltonian = jordan_wigner(fermion_hamiltonian)
qubit_hamiltonian.compress()
hamiltonian = cudaq.SpinOperator(qubit_hamiltonian)
num_qubits = hamiltonian.qubit_count
electron_count = molecule.n_electrons
1. Prepare an Approximate Quantum State¶
Start with a variational ansatz (e.g., from VQE) to approximate the ground state wavefunction on a quantum computer.
[4]:
if cudaq.num_available_gpus() > 0 and cudaq.has_target("nvidia"):
cudaq.set_target("nvidia")
else:
print("CUDA or GPU support is unavailable. Running with CPU simulator. Performance may be significantly reduced.")
cudaq.set_target("qpp-cpu")
[5]:
import numpy as np
@cudaq.kernel
def kernel(thetas: list[float]):
qubits = cudaq.qvector(num_qubits)
# Hartree-Fock
for i in range(electron_count):
x(qubits[i])
# UCCSD
cudaq.kernels.uccsd(qubits, thetas, electron_count, num_qubits)
parameter_count = cudaq.kernels.uccsd_num_parameters(electron_count, num_qubits)
exp_vals = []
def cost(thetas: list[float]):
exp_val = cudaq.observe(kernel, hamiltonian, thetas).expectation()
exp_vals.append(exp_val)
return exp_val
# Initial variational parameters
np.random.seed(15)
x0 = np.clip(np.random.normal(0, np.pi, parameter_count), -np.pi, np.pi)
optimizer = cudaq.optimizers.COBYLA()
optimizer.max_iterations = 300
optimizer.initial_parameters = x0.tolist()
energy, opt_params = optimizer.optimize(parameter_count, cost)
2 Quantum Sampling to Select Configuration¶
Measure this quantum state multiple times to obtain bitstrings, which correspond to Slater determinants (electronic configurations).
[6]:
sample_result = cudaq.sample(kernel, opt_params)
print(sample_result)
{ 01011010:1 11100100:2 11110000:997 }
3. Classical Diagonalization on the Selected Subspace¶
Construct a truncated Hamiltonian matrix using only the selected determinants. Diagonalize this matrix classically to obtain improved energy estimates.
[7]:
def pauli_element(
operator: cudaq.SpinOperator, b1: str | np.ndarray, b2: str | np.ndarray
) -> complex:
"""
Compute an inner product <b1|operator|b2>.
"""
if isinstance(b1, str):
b1 = np.array([i == "1" for i in b1], dtype=bool)
if isinstance(b2, str):
b2 = np.array([i == "1" for i in b2], dtype=bool)
num_qubits = hamiltonian.qubit_count
num_terms = hamiltonian.term_count
bsv = np.zeros((num_terms, 2 * num_qubits), dtype=bool)
coeffs = np.zeros(num_terms, dtype=np.complex128)
for i, term in enumerate(operator):
coeffs[i] = term.evaluate_coefficient()
bsf = term.get_binary_symplectic_form()
n = len(bsf) // 2
for j, e in enumerate(bsf):
if j < n:
bsv[i, j] = e
else:
bsv[i, j - n + num_qubits] = e
x = bsv[:, :num_qubits]
z = bsv[:, num_qubits:]
# b1 ⊕ b2 must match x for nonzero contribution
delta = np.bitwise_xor(b1, b2)
match = np.all(x == delta, axis=1)
if not np.any(match):
return 0.0
matched_x = x[match]
matched_z = z[match]
matched_coeffs = coeffs[match]
# i^{x·z}
x_dot_z = np.sum(np.bitwise_and(matched_x, matched_z), axis=1) % 4
i_phases = np.array([1, 1j, -1, -1j], dtype=np.complex128)[x_dot_z]
# (-1)^{b1·z}
b1_dot_z_parity = np.sum(np.bitwise_and(b1, matched_z), axis=1) % 2
sign_phases = np.where(b1_dot_z_parity == 0, 1, -1)
result_terms = matched_coeffs * i_phases * sign_phases
return np.sum(result_terms)
[8]:
import scipy
bitstrs = list(k for k, _ in sample_result.items())
subspace_dimension = len(bitstrs)
values = []
row_ids = []
column_ids = []
for i in range(subspace_dimension):
for j in range(i, subspace_dimension):
elem = pauli_element(hamiltonian, bitstrs[i], bitstrs[j])
if abs(elem) > 1e-6:
values.append(elem)
row_ids.append(i)
column_ids.append(j)
if i != j:
values.append(elem.conjugate())
row_ids.append(j)
column_ids.append(i)
values = np.real_if_close(values)
truncated_hamiltonian = scipy.sparse.coo_array(
(values, (row_ids, column_ids)), shape=(subspace_dimension, subspace_dimension)
)
# eigsh requires k < N; fall back to dense solver for small subspaces
if subspace_dimension > 1:
eigvals, _ = scipy.sparse.linalg.eigsh(truncated_hamiltonian, 1, which="SA")
else:
eigvals = scipy.linalg.eigh(truncated_hamiltonian.toarray(), eigvals_only=True)
5. Compare results¶
[9]:
## QSCI
eigvals[0]
[9]:
-2.1033232756074405
[10]:
## FCI Energy
molecule.fci_energy
[10]:
-2.14355458044603
Reference¶
[1] “Quantum-selected configuration interaction: classical diagonalization of hamiltonians in subspaces selected by quantum computers” K. Kanno, M. Kohda, R. Imai, S. Koh, K. Mitarai, W. Mizukami, and Y. O. Nakagawa (2023) arXiv: 2302.11320